"Hi, I see you added a note to Ibero-America regarding the Catalan-speaking population of Spain. This is of course correct, but I think that it is going to be problematic to try to classify the countries by language because several countries (including, but not limited to Spain) are multi-lingual. There's also Guarani, English, Nahuatl, Euskera, Gallego, etc... So I suggest getting rid of the language classification altogether (or at least try show the languages in a more general way that doesn't give the impression that there is only one language per country). What do you think?"
I did so because there are three sections in the article Ibero-America, as you can see: Portuguese-speaking, Spanish-speaking and Catalan-speaking. I thought it was good to add the note next to the Catalan-speaking one to say that the Catalan is also spoken in Spain. Other languages such as Guaraní would not fit in what I did, because we don't have a section called Guaraní-speaking. We neither have a section called Basque-speaking, in which "New Basqueland" and "The Independent Basque Country" are listed. If we had this situation, we could add a note that said: "hey, in Spain the basque is also spoken". But since the basque-speaking peoples do not have an independent state, they do not appear as an independent section in the Ibero-America article, so we don't mention them. But as we already have a section for the catalan-speaking countries (Andorra), it's good to add the note I added. I'm not sure if you understand what I mean xD Onofre Bouvila 17:20, 13 April 2007 (UTC)[reply]
I have reverted your deletion on List of publications in chemistry and put that and the new addition you added up for debate on Talk:List of publications in chemistry. That is how things are done. See the guidelines we agreed some weeks ago on the talk page. I have recommended deletion of the external link and keeping your excellent new addition. Please keep contributing to this page. Regards, --Bduke 22:15, 6 February 2006 (UTC)[reply]
No problem, thanks. Itub 22:39, 6 February 2006 (UTC)[reply]
SuggestBot picks articles in a number of ways based on other articles you've edited, including straight text similarity, following wikilinks, and matching your editing patterns against those of other Wikipedians. It tries to recommend only articles that other Wikipedians have marked as needing work. Your contributions make Wikipedia better -- thanks for helping.
If you have feedback on how to make SuggestBot better, please tell me on SuggestBot's talk page. Thanks from ForteTuba, SuggestBot's caretaker.
P.S. You received these suggestions because your name was listed on the SuggestBot request page. If this was in error, sorry about the confusion. -- SuggestBot 13:36, 13 August 2006 (UTC)[reply]
I left a message at the La Cucaracha's talk page regarding the wording of the lyrics, and provided some sources at least I consider more important than the normal amateur site. It went unanswered for more than two weeks before I decided to take action and change the las phrase. You jumped to revert my edits, leaving no reference of the "las dos patitas de atras" version nor any message at the talk page. What's more, you claim that to be the "real" version, providing no sources. I feel like reverting back to my version, but will let you explain youractions and source your position. I'll past this also at the article's talk page. Mariano(t/c) 08:29, 14 September 2006 (UTC)[reply]
I had reverted it because I guess something had gone wrong with the coding and the page was deleted. I tried refreshing the ñ page and it kept givnig me an error that it no longer existed, so I reverted it and it came back. Sorry, not hard feeling to whatever change you had made. --Speakslowly 23:17, 19 September 2006 (UTC)[reply]
Thanks for your message about this article. I've expanded it. Take a look and improve as necessary. Thanks, Scientizzle 22:05, 21 September 2006 (UTC)[reply]
Wow, that was a great expansion! Itub 23:02, 21 September 2006 (UTC)[reply]
Thanks for your note. I'm not sure what the problem is either, but I'll put the word out for some help. Thanks again! -AED 21:57, 8 October 2006 (UTC)[reply]
Sorry about the flub on the conversion rate. I saw "currently" without any indication how current "current" was, and wanted to insert a date (and felt the need to validate that the rate hadn't changed since whenever "current" was). In case you're curious how I messed up: I got 9¢ (US) per peso backwards in my head. AdamRoach 18:51, 12 December 2006 (UTC)[reply]
Hi, I rewrote this article. I apologize if I seem to have lost some of your wording. You got this article off to a great start, and I could not resist. Feel free to revise what I put up. I will insert a figure soon. --Smokefoot 18:14, 28 December 2006 (UTC)[reply]
Thanks, I've commented about the figure in the article's talk page. The only thing I miss is the mention of the equivalent representation in terms of resonance structures. I may add it later, but it sorely needs a figure too. :) Itub 20:33, 28 December 2006 (UTC)[reply]
Thank you for your patience (with my invasiveness) and guidance.--Smokefoot 20:42, 28 December 2006 (UTC)[reply]
No problem, it's not like I own the article, I didn't even start it! The article is better now thanks to your contributions too. Itub 21:12, 28 December 2006 (UTC)[reply]
Hi, I notice that you are interested in molecular orbitals. Thus, for you to note, I have just started the molecular orbital theory article if you are interested in contributing. Thanks: --Sadi Carnot 23:16, 2 January 2007 (UTC)[reply]
Could you have a look at the energy and the last two edits by Hallenrm?[edit]
Either I've completely lost my mind, or I'm dealing with an invariably insulting editor who has an advanced degree in chemistry, yet really doesn't understand key parts of the physics of the subject. So, I'm asking for some badly needed RfC on the chem section of this article. I'm leaving an identical message for user:smokefoot and everybody else with a chem degree I can think of. Gracias for the outside view. SBHarris 09:34, 19 January 2007 (UTC)[reply]
I'm puzzled by your removal of the merge tag from non-covalent bonding with the reason "non-covalent bonding is important in the field of supramolecular chemistry according to Lehn". I'm not questioning the importance of non-covalent bonding. What I'm questioning is whether the Wikipedia articles currently titled non-covalent bonding and intermolecular interaction actually deal with different topics. My opinion is that they don't, and therefore having two articles is redundant. I don't have a strong preference regarding the title of the resulting article, though. --Itub 12:33, 12 March 2007 (UTC)[reply]
While I appreciate your effort to organize, I have interest in supermolecular chemistry and although one could loosely merge these without to much difficulty, I reason that they should remain separate. Although I need to read more in this area, from what I understand so far Jean-Marie Lehn, the main founder of this field, who won the 1987 Nobel Prize for his work in SMC, defines “molecular chemistry”, as begun by Friedrich Wohler in 1828, as related to the making and breaking of covalent bonds. He contrasts this with the new field of “supramolecular chemistry” as related to the making and breaking of non-covalent bonds between supermolecules. Intermolecular forces, on the other had, is a more general term which may include the covalent bond if fashioned so. If you don’t believe me, look at this edit by User:V8rik, in which the intro definition of supramolecular chemistry is defined by the use of the term “noncovalent bonding. I hope this helps? --Sadi Carnot 00:26, 13 March 2007 (UTC)[reply]
Hi there! Just curious, why did you revert the numerous contributions made by James? —Signed, your friendly neighborhood MessedRocker. 11:10, 15 March 2007 (UTC)[reply]
Just thought i'd mirror what i posted to MessedRocker's page;
I've just spoken to him (James) on the phone and he's informed me that his account has been re-infiltrated again by whoever keeps using his laptop, most likely his son. He's been away for 3 days on a trip, so it's unlikely any of the things that were added were from him directly, and he wanted to explain also that he's looked at his page and will change it when he gets back.
He also informed me that his previous account, User:J.Spudeman was similarly infiltrated and the password and email changed so he was unable to recover it. Regardless, i'll see him on saturday morning and speak to him then, but i just wanted to give you the "heads up". Cheers, J O R D A N[talk ] 16:08, 15 March 2007 (UTC)[reply]
Thanks for letting me know. --Itub 19:19, 15 March 2007 (UTC)[reply]
Do you have a list of all X-center, Y-electron bonds somewhere? I made a new template at Template:Chemical bonds and would like to add them to it. --HappyCamper 12:27, 16 April 2007 (UTC)[reply]
I don't have a list, but I just added delta bonds, which were missing from the template. --Itub 12:30, 16 April 2007 (UTC)[reply]
I haven't decided what to put in that template yet, since I wanted to avoid duplicating the items that are found in Category:Chemical bonding. And of course, you're always welcome to edit my templates. :-) By the way, if you have a picture of a particle in a box, that would be really nice...do you have access to a lab, where you could say, heat a cubic crystal of NaCl in some sodium vapor? --HappyCamper 17:27, 23 April 2007 (UTC)[reply]
Nope, I'm a theoretical/computational chemist. I tend to break things when I go into a lab. ;-) --Itub 17:48, 23 April 2007 (UTC)[reply]
Thanks for all your help on Francium. Your constant input on the peer review, your support on the FAC, your solution to the solution problem, and your edits to the article itself are all greatly appreciated. If you ever have an article being peer reviewed (and I'm not already making comments there), let me know and I'll settle up the score. --CrypticC62 · Talk 18:21, 29 April 2007 (UTC)[reply]
Ivan, let me clarify my position. It's not that I object to anyone editing the article, it's the fact that it has been done so badly. Let me give a few examples. You can still see the original at user:petergans/sandbox
Introduction: Adding a catalyst will fasten the reaction!
The expression for forward reaction rate has been separated from the defining equilibrium expression.
"Despite the failure of this derivation, the equilibrium constant for a reaction is indeed a constant, independent of the activities of the various species involved "!
Contradictory definitions
______________________
αA + βB σS + τT
...
...
The equilibrium constant K is defined as
where [A] represents the chemical activity of the species A at equilibrium composition. Often (conditions are given below) these activities can be approximated by molar concentrations or partial pressures, giving the easier to measure and use constants Kc and Kp encountered in high-school chemistry courses.
_______________________
This last statement is fundamentally unsound and is one of the reasons why I undertook a re-write. In media of constant ionic strength there is no such approximation. Even Atkins may be unaware of this, but I can't say for sure as I only have the 1st. Edition (being the old inorganic chemist that I am).
"What this means is, the time derivative of the free energy vanishes, signalling a stationary point. This derivative is usually called, for certain hisorical and technical reasons, the free energy change[2]" What rubbish! he's quoting out of context. Even when one replaces the nonsensical time derivative by derivative with respect to the reaction coordinate it still does not make sense to equate a derivative with a free energy change. There's a real dilemma here: equilibrium is taught through kinetics in schools and through thermodynamics at University. I really object to schoollchildren being told "this theory is wrong but it gives the right answer".
"The most basic approach is to manipulate the various equilibrium constants till the desired concentrations are expressed in terms of measured equilibrium constants (equivalent to measuring chemical potentials) and initial conditions" again rubbish as it stands. If we say manipulate various expressions relating to the equilibrium, that's getting nearer to being meaningful, but the idea of measuring chemical potentials in this context defeats me.
He's pasted this in from the old article, but the notation is inconsistent with the notation given earlier. The same applies to the rest of the section.
It was for these reasons that I said his editing was irresponsible and wanted to start again from my original. It's hardly my job to correct his errors is it? I should be grateful if you would post some conciliatory remarks on the chemical equilibrium talk page - if the article is reverted to the original I will quit editing it. Like you say, I put a lot of time and effort into the revised version. I did it because I wanted to get the science right and reverting to the original will mean that the science in the article is not right. There's no way I could put it right by an series of incremental edits such as I have done, for example, with solubility product.
May I add that I declined to enter the entropy of mixing "debate" though in fact I initiated it with a remark I made to happy camper. You were, of course, right, but loom91 would not accept your arguments. His statement "Actually, it's more general to talk about entropy, because it is a more fundamental and universal quantity than Gibbs energy" shows him to have a poor grasp of thermodynamics.
All right, but I'm going away for a few days and I won't have time to do that until Monday. Please be patient, the world won't end if the article is not perfect for a few days. --Itub 06:47, 18 May 2007 (UTC)[reply]
The content is identical with that of equilibrium constant, so I am happy to replace the content by a redirect, or you do it if you agree. Petergans 10:08, 22 May 2007 (UTC)[reply]
Ok, I've turned it into a redirect. I hadn't noticed that the content was identical, so I thought that a more involved merge would be needed. --Itub 12:37, 22 May 2007 (UTC)[reply]
Interesting point about reactions going to completion. I've done the maths and have come up with a semi-quantitative measure. For the reaction A+BAB, if one starts with 1 mol of A and 1 mol of B reaction "goes to completion" when Lg K > 2 lg N, where N is Avagadro's constant: for this reaction lg K > ca. 47.5/mols of reactant. This translates to Go > ca. 118 kJ mol-1. I doubt if this result is original. If we can find a citable source it might be worth adding it as footnote. Petergans 14:15, 23 May 2007 (UTC)[reply]
I've looked at bit at J. Chem. Ed. and couldn't find an article with this type of calculation, but I wouldn't object to adding it as a footnote as it is a relatively straightforward derivation from the equations that are already in the article. However, when I tried to do this calculation I got 271 kJ/mol. Are you sure you didn't mix up the log10 with the ln? --Itub 17:23, 23 May 2007 (UTC)[reply]
You are right, I forgot the 2.303. Please put this stuff in yourself if you want to, as I won't touch the article again for a while.
Here is my derivation.
TA=[A]+K[A][B]
TB=[B]+K[A][B]
When TA=TB [A]=[B], so TA=[A]+K[A]2
Since [A}<<TA, TA=K[A]2. Putting TA=1 and [A]=1/N, the result follows. Footnote or mini section? I have no preference. Petergans 19:24, 23 May 2007 (UTC)[reply]
The result can be somewhat generalised. For A+nBABn
TA=[A]+K[A][B]n
TB=[B]+nK[A][B]n
Assuming that TB=nTA it comes out that the criterion is Lg K > n+1 lg N for 1 mole of B. Petergans 08:01, 24 May 2007 (UTC)[reply]
See answer on my TALK page. And, BTW, sorry for mistakenly posting this first on your user page, rather than properly your TALK page! A breach of wiki-ettiquette I know better than! SBHarris 21:25, 14 May 2007 (UTC)[reply]
Hi, I was thinking that some discussion of the alchemical principles (salt, sulphur, mercury) would be useful to add to the history section of chemical element, or perhaps to history of the molecule. However, I'm not that familiar with this topic, and since the ancient history of chemistry seems to be one of your favorite topics I thought you might know more about it. Cheers, Itub 08:01, 24 May 2007 (UTC).[reply]
Yes, I already have most of this typed up in a sub-section called "Heat and affinity" of a chapter called "Affinity and Free Energy" (chapter 11, pgs. 329-368) of a new 2007 textbook on Human Chemistry[1] publishing soon. I'll just give you the 7-page subsection here: Heat and affinity with the 25-references included and I'll leave it to you to format (short on time presently). Later: --Sadi Carnot 11:02, 24 May 2007 (UTC)[reply]
I'm currently trying to puzzle out whether the catalyst in the Haber process#catalysts speeds up the reverse reaction or not not. Looking at the proposed mechanism, it seems very unlikely to me. One can imagine all sorts of free radical reactions occurring rapidly in the gas phase once the first N-H bond is thermally homolysed. I have two questions for you. 1) Do you know of any literature on the thermolysis of ammonia? Have the dissociation energies (for loss of one hydrogen atom) of NH3, NH2 and NH: been calculated ab initio? If not, would you like to amuse yourself with some juicy open-shell calculations? The point at issue is whether a catalyst always speeds up both forward and backward reactions when the mixture is not at equilibrium. Petergans 16:44, 28 May 2007 (UTC)[reply]
I don't know the literature about the thermolysis of ammonia, but I'm pretty sure that the dissociation energies have been calculated. I don't see why the catalytic mechanism for the Haber process wouldn't speed up the reverse reaction, and I don't know any exceptions to the principle of microscopic reversibility. It is true that there could be side reactions involving NH and NH2, but that is true for both the forward and the reverse reactions. However, I don't imagine that these reactions will be of major importance, because they would presumably be reversible too and lead to species that are less stable than N2, H2, and NH3. The fact that the Haber process works leads me to think that, but I'm not an expert on it. --Itub 07:13, 5 June 2007 (UTC)[reply]
for a multi-stage reaction. It clearly illustrates that, for example, in the carbonic anhydrase- catalysed reaction of CO2 with H2O the catalyst affects the activation energy (TS1) for the forward reaction, but not neccessarily for the reverse (TS2). It will only affect the reverse reaction rate if it happens to be also the rate-determining step and if the reverse reaction must follow the same reaction path. Petergans 18:17, 16 June 2007 (UTC)[reply]
It's not that simple. The concept of rate-determining step has its limitations,[1] but in any case for this particular figure the RDS for both directions is TS2. If you solve all the differential equations you'll see that the rate will change equally for both directions, regardless of which TS you modify. Also, if you could change the rate in only one direction, you would be changing the position of equilibrium. While it is true that K=k1/k-1 is not generally valid, it is true that the rates in both directions are equal at equilibrium. Therefore if you changed one without changing the other you would affect the equilibrium.[2] For more information about the principle of microscopic reversibility, see [3]. The only exception to this principle I know of is in the case of photochemically activated reactions; in those, after the excitation the system finds itself in a different potential energy surface, which usually goes downhill to the products. --Itub 09:07, 17 June 2007 (UTC)[reply]
I'm not expert in this field, so I'll accept what you say about microscopic reversibility. However there are a couple of matters that are still not clear.
I think the activation energy for the first step is not ΔG1, but the difference between C and TS1.
The equilibrium constant depends on the energies of C and D independently of intermediate energies. The catalyst affects the energy of an intermediate state, but not the equilibrium composition of the mixture, so in this multi-step situation one reaction can be speeded up without affecting the final outcome. The rate of reaction away from equilibrium conditions is not the same as that at equilibrium.
Why are the energies of C and D shown as minima rather than constant values?
What about catalysis by enzymes? If you broadly accept the priciple that enzyme and substrate are like lock and key an enzyme will only speed up the forward reaction. As an example, consider carbonic anhydrase, CO2 + H2O HCO3- + H+. It works by a) polarising a water molecule by coordination to Zn, b) bringing CO2 into close proximity of the Oδ- attached to Zn and c) having a basic site handy to accept the liberated proton. I don't see how the enzyme can adapt to accept a bicarbonate molecule and a proton rather than expel them. Petergans 20:06, 19 June 2007 (UTC)[reply]
I'll use the same numbering for clarity.
I'm assuming you are talking about the process C -> A -> B -> D. In this case, the first step has a barrier with the difference TS1−C. However, in the context of the system discussed in the Curtin-Hammett article, the "first" step is either A -> C or B -> D. It is assumed that A and B are always in equilibrium with one another, and that C and D are not, because the barriers from C to A and from D to B are insurmountable under the reaction conditions (i.e., have a half-life longer than hours, months, years, or billions of years, etc. depending on the interests of the experimenter).
It is true that the equilibrium constant depends on the energies of C and D independently of intermediate energies. However, the equilibrium is dynamic, which means that for equilibrium C <-> D, for example, C is being converted into D exactly at the same rate as D is being converted into C. From mass balance it follows that whatever happens to the overall rate for C -> D has to be compensated by an equivalent change in the rate for D -> C. Think about what would happen if you added a catalyst to a system that is already in equilibrium, and the catalyst only affected the rate in one direction.
That's a common drawing convention used in physical organic chemistry to indicate that those states (reactant, product, or intermediate) are in fact minima in the (multi-dimensional) potential energy surface. However, it is just for illustration, and some authors choose to use other illustrations, such as straight horizontal lines. The minima as plotted only have a strict meaning when the reaction coordinate is well-defined, which can only be plotted for very simple reactions. I'm using the same style as this book, for example: [4].
Enzymes can certainly catalyze the reverse reaction as well, and many reactions of metabolic interest are reversible and need the enzyme to catalyze it both directions (I think the hydrolysis of CO2 is a good example, due to its a role in the regulation of pH of the blood). At least the book I have available, Lehninger's Principles of Biochemistry, 2nd ed. clearly states that enzymes also catalyze the reverse reaction. Although there is still some controversy regarding the best explanation for enzymatic catalysis, which can also vary from enzyme to enzyme, it is pretty clear that it is not by simply binding to the substrate as per the lock-and-key model. A more accepted explanation is induced fit, which means that the enzyme binds to the transition state better than to the substrate itself, thus lowering the effective barrier. --Itub 11:06, 20 June 2007 (UTC)[reply]
Many thanks for these comments. I'm still not completely convinced, but I don't want you to spend any more time on this discussion. I think there are two situations which need to be considered: at equilibrium forward and backward rates are equal, but away from equilibrium this is not so. The catalyst is mainly effective away from equilibrium. I find it difficult (but not impossible) to accept that at equilibrium the catalyst affects both reaction rates equally. It's also possible that at equilibrium the catalyst has no effect. The kinetics of multi-step reactions is not something that we should get into, is it! Let's leave it at that. Petergans 14:25, 21 June 2007 (UTC)[reply]
One last thought, after all. If the catalyst simply lowers the energy barrier to the point where the forward reaction proceeds at its "natural" rate equilibrium will be reached quickly. In the ammonia case the barrier is the dissociation of N2; once this process is speeded up by the Fe catalyst the reaction proceeds "normally". The back-reaction is unaffected. Petergans 09:46, 22 June 2007 (UTC)[reply]
It has to be affected, due to microscopic reversibility. I think you are focusing too much on the dissociation of N2, which admittedly is a highly unfavorable step which hinders the reaction. But the reverse reaction also faces quite a big barrier. Although I don't know exactly what it is because I don't know the mechanism well, the barrier is possibly even larger than that for the forward reaction, given that the formation of ammonia is slightly endothermic (DeltaHf = -10 kcal/mol). Ammonia is very stable (in the kinetic sense) at room temperature; one does not see it decomposing into N2 and H2. But if you put ammonia in a Haber reactor you should see it decompose until it reached equilibrium, and it would reach it faster thanks to the catalyst. --Itub 11:16, 22 June 2007 (UTC)[reply]
The reverse reaction must begin with . The energy of this dissociation is much less than for the dissociation of N2. Once these radicals start flying around chain reactions will occur. The radicals involved will be the same as in the forward reaction, but it's easy to see how nitrogen atoms can be produced in the back-reaction without the need for a catalyst. I'm still troubled by the idea that at equilibrium the catalyst affects both overall reaction rates in such a way that the ratio remains constant. I can see it in principle for a reaction coordinate with a single maximum, whose height is affected by the catalyst, but not for multi-step systems.Petergans 18:44, 22 June 2007 (UTC)[reply]
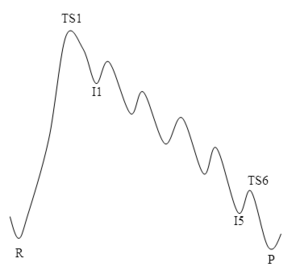
I've made this sketch of a hypothetical six-step mechanism to illustrate a point. In this sketch, the rate in the forward direction is determined by the difference TS1-R, and in the reverse direction by TS1-P. The rate-determining step is the same in both directions. One could think that the reverse mechanism would be easier because it is only one small barrier at a time instead of one huge barrier, but sadly that's not the case. Imagine you start from P, and "climb" one step to I5. Once you are in that state, you are faced with two possibilities: climbing one more step to I4, or going back to P. Unfortunately, going back to P is much more likely because the barrier in that direction is lower. Effectively what happens is that you are establishing an (unfavorable) equilibrium between P and I5 before you can even climb the second step. And so on, until you reach TS1 and finally "fall down" to R.
Back to the Haber process, let's assume for a moment that the first step is the dissociation of N2, N2 -> N + N. The reverse step would be the formation of N2, N + N -> N2, a step which might even be barrierless, but there had to be a large series of steps that had to be climbed starting from ammonia, something like 2NH3 -> 2N + 6H. Note: I don't think that's the real mechanism, but rather a free radical mechanism as you suggest. That means that you wouldn't need to fully dissociate N2 as the first step, you could have something like N2 + H -> N2H and so on. Also note that in the catalytic mechanism you are not really lowering any of these steps but rather undergoing a completely different mechanism, with more steps, the first of which is probably the adsorption of N2 and H2 on the surface of the catalyst. --Itub 18:47, 23 June 2007 (UTC)[reply]
Now we have come full circle. I agree that the catalytic mechanism probably involves the adsorption of N2 and H2 on the surface of the catalyst. The article on the Haber process suggests that both molecules are dissociated on the Fe surface. What is certain is that the activation energy for breaking the N-N bond is lowered. Now consider the reverse reaction. I can imagine that nitrogen atoms are produced by such reactions as NH2+H NH+H2 and NH+H N+H2. If this is the case, then the dissociation of ammonia at the temperature and pressure of the Haber process can reach equilibrium rapidly without the aid of a catalyst because there will be a steady state concentration of nitrogen atoms and radicals, including NH, NH2. The forward reaction rate, in the absence of a catalyst, is slow. I agree that H atoms may well be present but the evidence suggests that they don't have enough energy to kick-start the reaction. Whilst the catalysed reaction may well obey the principle of microscopic reversibility it is likely that the catalyst has no effect on the dissociation process. That was my original concern. If NH3 or an intermediate is adsorbed by the catalyst that would be yet another mechanism.
P.S. An edit war has broken out in regard to the Law of Mass Action. The original article was totally useless, so I set about reading the papers by Guldberg and Waage and produced the version of 13.45, 24 June, based on these readings. Loon91 immediately reverted most of it without explanation. An issue here is that many textbooks give a definition of the law which is significantly different from the one developed by G&W, so it's no use citing those sources to support such a definition. I guess you would not want to get involved, but this guy is a loose cannon and I don't know how to deal with such hostility. Petergans 11:46, 25 June 2007 (UTC)[reply]
Unfortunately I don't know much about the history of the law of mass action, and I don't think I want to get involved at this point. The only thing I can suggest is to discuss the topic calmly and try to find good recent references about it. If the textbooks are wrong, I'm sure someone must have written an article on J. Chem. Ed. trying to clarify the issue. (Frankly, I don't even understand what the issue is. This is a historical topic about which I really know next to nothing.) --Itub 12:22, 25 June 2007 (UTC)[reply]
I have at last obtained pdf's of the two articles you cited, and have read the one by Clugston carefully. There is no doubt that entropy maximisation and free energy minimisation are two sides of the same coin. In that sense Clugston's article is "trivial". Moreover I detect something that looks like a circular argument: where do the equilibrium concentrations come from? Put another way, why are the dissociation constants for ethylacetate and propylacetate different? The entropy argument is the same for both, so it must be mainly the difference in standard free energy between reactants and products that makes equilibrium constants different and hence, determines the composition of an equilibrium mixture. I just don't like separating one entropy effect for all the others such as those involved in solvation/desolvation about which we know next to nothing. Petergans 21:06, 31 May 2007 (UTC)[reply]
Re-read Clugston's paper. He acknowledges that the argument does not apply to heterogeneous equilibria. That's solubility out of the window! The statement that standard free energy change refers to pure substances is not correct in general. For gases and liquids the standard states are the pure substances, but this is not true of solutes; I don't think that the argument can be applied to solutions because there are entropy factors that apply to the solvent itself, particularly in reactions where ions are involved. I conclude that it needs to be stated that the entropy of mixing idea does not apply universally. Petergans 10:18, 1 June 2007 (UTC)[reply]
I think it would be good to qualify that the entropy of mixing does not apply to heterogeneous systems, which are in some ways similar to phase equilibria. It is also true that the ideal entropy of mixing is not exact for non-ideal solutions, given the entropy changes in the solvent that you mention. That said, I still think the entropy of mixing is important because it is the only contribution to the free energy that explains the existence of chemical equilibrium for ideal systems. If it is excluded, a plot of free energy vs advance of reaction has no minimum but is a straight line, and one would expect 100% conversion to the products (assuming they have a lower free energy than the reactants). The entropy of mixing is a curve with a minimum at 50%. When you add this curve to the straight line due to the free energy of reaction, the position of the minimum shifts depending on the magnitude of the free energy change. (As we discussed above, if the free energy change is large enough, the minimum can become negligible, resulting in a practically 100% equilibrium position.) In other words, the entropy of mixing "causes" equilibrium by discouraging having pure products, but it is indeed the free energy of reaction that determines the value of the equilibrium constant, as is apparent from the equation DeltaG0 = -RTlnK. --Itub 06:58, 5 June 2007 (UTC)[reply]
Thanks for these comments. I, too have been away. To be perfectly general we also need to explain the existence of a minimum in in the case of heterogeneous equilibria. I haven't given this any thought, as I'm not a thermodynamicist. Do you know if anything has been published on this subject? Petergans 18:06, 11 June 2007
(UTC)
There are several proofs of the existence and uniqueness of chemical equilibrium cited in Van Zeggeren and Storey, The computation of chemical equilibria (1970). Unfortunately they are conference reports rather than publications. D.B. Shear, J. Chem. Phys. (1969) 48, 4144-4147 appears to have a proof of uniqueness. I suspect that these proofs might be difficult to convey to the general reader. Incidentally, the entropy of mixing for ideal gases is also discussed and cites M.W. Zermanski, Heat and Thermodynamics (1957) so that idea is not very new, even if it has only recently crept into Atkins's Physical Chemistry. Petergans 14:21, 13 June 2007 (UTC)[reply]
Well, the entropy of mixing was known by Gibbs, at least according to the mixing paradox article; I wouldn't be surprised if he had already considered its role in equilibrium in one of his famous papers published around 1870. But I haven't read any of Gibbs's papers so I can't tell for sure. --Itub 14:29, 13 June 2007 (UTC)[reply]
As noted, Hallen, after totally screwing up the energy article, is now going to actively prevent anybody from writing a summary of science-based energy. I really see no alternative but to get him banned from editing the thing. Otherwise we're never going to be left alone to write it. I'm open to suggestions.SBHarris 21:50, 11 June 2007 (UTC)[reply]
I recently added a paragraph to the article on the anomeric effect, in which I pointed out that there are actually two conformations of the equatorial 2-tetrahydropyranyl ether that MUST show n-sigma* effects. Anyone with a molecular model of this molecule can see this. Yet you deleted my contribution. Please explain. Vgsbox 11:38, 14 June 2007 (UTC)[reply]
You really have NOT given me any reason for your desire to delete the article on STR3DI32. I can only assume that you are in opposition to any method of molecular modeling that is not orbital based. Am I correct? Is this move just another self-appointed censoring? Vgsbox 11:38, 14 June 2007 (UTC)[reply]
Please read WP:N, WP:COI, WP:NPOV, WP:SOAP, and WP:TEND. If after that you still don't understand the reason, I cannot help you. --Itub 11:48, 14 June 2007 (UTC)[reply]
Besides STR3DI32, there are only a few, if any, molecular mechanics based programs that can be described as quantitative versions of the VSEPR theory. Since the VSEPR theory is, and will remain, an invaluable tool in organic chemistry, an article on STR3DI32 is also valuable and important. None of the principles that you refer to, concerning articles in the Wiki, have been violated. Arev you simply trying to prevent other readers from knowing about the existence of a MM program that quantitates the VSEPR theory? Vgsbox 11:57, 14 June 2007 (UTC)[reply]
Please take your arguments to Wikipedia:Articles for deletion/STR3DI32, where other Wikipedia editors with no part in this dispute will help reach a consensus. --Itub 12:05, 14 June 2007 (UTC)[reply]
Can you explain why you redirected the article Energy (Chemistry) to energy. There was already a conflict with physchim62 regardingthe earlier redirect, in which Jrefree has given some suggestions. From the above post I notice that you somehow specialize in such acts. I really feel concerned with the wikipedia future, if people like you are given a free handHallenrm 04:11, 20 June 2007 (UTC)[reply]
Ok, I've followed one of Jreferee's suggestions and nominated the article for deletion. Cheers, Itub 08:03, 20 June 2007 (UTC)[reply]
Most of the literature points to 1873 not the original paper from 1870! But know I found a problem in furfural. The original publication of Döbereiner from 1832 states that he wants to introduce formic acid (Ameisensäure) for several medical issues and therefor introduces a synthesis from suggar. 1 Theil desselben in 2 Theilen Wasser ... mit 2½ bis 3 Theilen Manganhyperoxid... 3 Theile Schwefelsäure (1 part suggar in 2 parts water with 2½ to 3 parts Manganhyperoxide and 3 parts sulfuric acid at 60°C). There are no ants in this discription at all. The discription of furfural is small in this article and states: oil like substance smelling like almonds is a by product of the synthesis. I think we should change the article accordigly.--Stone 16:57, 27 June 2007 (UTC)[reply]
As far as I'm concerned, please go ahead! I don't know anything about the history of furfural so I have no reason to object. It does sound more plausible that it was obtained from sugar than from ants. --Itub 12:15, 28 June 2007 (UTC)[reply]
Thanks for uploading Image:Players handbook 2nd ed.jpg. The image description page currently specifies that the image is non-free and may only be used on Wikipedia under a claim of fair use. However, the image is currently orphaned, meaning that it is not used in any articles on Wikipedia. If the image was previously in an article, please go to the article and see why it was removed. You may add it back if you think that that will be useful. However, please note that images for which a replacement could be created are not acceptable for use on Wikipedia (see our policy for non-free media).
If you have uploaded other unlicensed media, please check whether they're used in any articles or not. You can find a list of 'image' pages you have edited by clicking on the "my contributions" link (it is located at the very top of any Wikipedia page when you are logged in), and then selecting "Image" from the dropdown box. Note that any non-free images not used in any articles will be deleted after seven days, as described on criteria for speedy deletion. Thank you. BetacommandBot 21:50, 10 July 2007 (UTC)[reply]
I saw your prod. I'm not sure if this is entirely made up by the first editor to this page since it has been mentioned elsewhere. Some chemical compounds are also rather imaginatively named (what about cubane or prismane?), so that in itself is not sufficient reason to say this name is made up. Perhaps you might want to change it to a db-not notable, but that is a problem too because the non-chemist mafia uses such reasoning to attempt to delete many of our chemical stubs.
Anyway, I have no vested interest in this article, but I do not believe wp:made-up is an appropriate reason. --Rifleman 82 09:15, 11 July 2007 (UTC)[reply]
Maybe I extrapolated WP:MADEUP in the sense that the name was not made up by the initial author of the article, but it is certainly a "made up" name in a more general sense. There are many imaginatively named molecules, such as cubane, where the names do get used in the "real world", and as such they will appear in real publications and indexed by CAS. But that is not the case with penguinone. --Itub 09:20, 11 July 2007 (UTC)[reply]
If I may add, I started looking for real information on this molecule precisely in an attempt to demonstrate its notability in case someone nominated it for deletion. To my surprise, I couldn't find anything, which lead me to the conclusion that it should be deleted. I'm all from saving chemical articles from "non-chemistry mafia" deletionists, which is why I worked on rescuing caesium perchlorate. But penguinone is a joke. If anyone has the inclination to read the 11 papers about 3,4,4,5-Tetramethyl-2,5-cyclohexadienone and can conclude that there is something relevant to say other than that some people think it looks like a penguin, they are more than welcome to create a real article under the chemical name. --Itub 09:39, 11 July 2007 (UTC)[reply]
I see your point, and I agree. There are practically no limits on the number of chemicals which can and do exist, but not all are "important" in the sense of them having a practical use as a catalyst, building block, etc, or even being of academic interest. --Rifleman 82 10:36, 11 July 2007 (UTC)[reply]
In this edit you put two D&D Player's Handbook covers into a gallery. Unfortunately book covers are only accepted under Wikipedia's Fair Use guidelines. Most importantly,
"Fair use images may never be included as part of a photo gallery...". I disagree with the policy, but it's still likely to get deleted by someone else. The additions may be useful, but to pass by the more strict enforcers of the fair use rules you'll need to break it out of the gallery and add some notes about why each cover is worth mentioning. (Some notes on the history and differences between the books might be enough.) — Alan De Smet | Talk 01:52, 12 July 2007 (UTC)[reply]
Ok, I've move them out of the gallery, but I had put them there only in an attempt to improve the layout of the page (the floating images were screwing up the headings and stuff like that). I also expanded the history section. --Itub 08:06, 12 July 2007 (UTC)[reply]
I see you're recatting a lot of stuff right now. I can help with User:Chem-awb, if you give me a list of what maps to what. So, would you like a hand? It would save you a lot of typing and clicking? --Rifleman 82 09:26, 6 September 2007 (UTC)[reply]
I'm done for now, but it would be good to know what format such a list should have, in case I need it in the future. Note that I'm not renaming categories, but in some cases removing categories that are too general, in others substituting them with a more specific category, and yet in others adding categories. --Itub 09:33, 6 September 2007 (UTC)[reply]
I'll have to key it into AWB myself anyway, so it doesn't matter. You can have something like:
I'm not wedded to this format. As long as I can understand what change you want to make, and the article titles you want to apply this change to, I can take care of the rest. --Rifleman 82 09:46, 6 September 2007 (UTC)[reply]
Could you please respond to the question addressed in the RFC: whether the country is ambiguous or not? I would like to close that RFC with some conclusion and start a different one on the topic that people have been writing about. Thanks! panda 06:01, 14 September 2007 (UTC)[reply]
Thanks for participating in the RFC: Country data in Nobel lists! Since you indicated that you agreed with item 3.1 (use common name) or 3.2 (use names shown on the Nobel site), could you please revisit the RFC and respond to the question in the section called Clarification & Questions related to 3.1 or 3.2? Thanks! –panda 04:37, 30 September 2007 (UTC)[reply]
I don't know if this changes anything but wanted to let you know that I've discovered what the country data is supposed to be. You can find it under the point Country data defined in RFC: Country data in Nobel lists. –panda 14:55, 10 October 2007 (UTC)[reply]
My opinion is that the article is ready for WP:FAC, but the GA was put on hold with comments that are so vague as to be nearly useless. I say forget about GA and go straight for FA. At least the process is more open, with more people giving their opinion, and with more dialog where one can generally ask for specific, actionable items and ignore the objections that are not clarified. --Itub 08:21, 18 October 2007 (UTC)[reply]
Thanks Itub. I think for the moment I'm going to ask for clarification on the GA review. It can't hurt to see what the concerns might be. There's no hurry, after all. I was planning to take it through for FAC once the GA was passed. — RJH (talk) 14:43, 18 October 2007 (UTC)[reply]
Smokefoot and I have been working on this page, and from my limited experience it seems to cover most of the common techniques. Do you have any comments about the style, formatting, etc.? Or, any suggestions for inclusion? Thanks for your comments--Rifleman 82 15:00, 18 October 2007 (UTC)[reply]
Sorry about that! I have no idea what happened there, must have inadvertently highlighted your paragraph and overtyped it — iridescent 14:09, 23 October 2007 (UTC)[reply]
I really like your point about Jekyll/Hyde. That issue is worth clarifying. - JehochmanTalk 14:18, 23 October 2007 (UTC)[reply]
I'm glad we agree about something! --Itub 08:45, 24 October 2007 (UTC)[reply]
Physchim62 posted a bunch of evidence against me. I really didn't want to expand the case, but he's forced me to respond. I hope you understand that this isn't personal. Our conversations were going so nicely. Sigh. - JehochmanTalk 17:16, 1 November 2007 (UTC)[reply]
Good catch on that AfD - I saw the very badly written article which was posted last night and reacted a bit too hastily. Thanks for taking the time to confirm that there was a worthwhile subject there. I've withdrawn the nom, and rewritten the article from scratch with the references you found. Iain99Balderdash and piffle 17:49, 6 November 2007 (UTC)[reply]
No problem, and you did a great job on the rewrite! The article originally didn't have much that was salvageable, so deleting it would have been understandable. --Itub 10:11, 7 November 2007 (UTC)[reply]
You asked on my talk page if I knew where the symbol "R" comes from, when used to symbolize any arbitrary hydrocarbon radical in a structural formula. I don't know the historical origin of this symbol, but I can tell you that it is either very rare or not found at all among chemical formulas used in the nineteenth century. I suspect it was first used in the early or mid-twentieth century. I have always confidently assumed that the person who introduced it (whoever that was) chose the letter R to stand for "radical"; but that is, as I say, just an assumption. I'm sorry I am not able to give you a better answer to this question. Ajrocke 16:21, 7 November 2007 (UTC)[reply]
I find it incredulous that one would attack even the man who invented the term 'folklore' and started the field of 'supercentenarian tracking.'
Such mass hysteria seems to be working. User BHG is one of the top-10 editors by edit count on Wikipedia and has lots of friends. I can't imagine most WWI veteran articles surviving if this trend continues. I do urge everyone who considers tracking the oldest-old, whether WWI vets or no, to chime in on these debates.
Sincerely, Robert YoungRyoung122 17:08, 9 November 2007 (UTC)[reply]
Thanks for doing the right thing on that user page. I had forgotten about that page -- I had meant to mark it for deletion it weeks ago. Your prompting has reminded me to do that. Karl Hahn (T) (C) 16:28, 12 November 2007 (UTC)[reply]
Good work on this article. Have you seen the new lead to computational chemistry? I think it is wrong, but I do not really have time until tomorrow to address it again. --Bduke 09:39, 13 November 2007 (UTC)[reply]
It is wrong, or at least WP:UNDUE. When was the last time you saw even a mention of mathematical chemistry in a computational chemistry text or review? ;-) I've reverted the last edit. --Itub 10:08, 13 November 2007 (UTC)[reply]
Perhaps you should explain to User:NYScholar your original reason for including the Nobel Prize in Economics article in WP:LAME on that user's talk page. I'm pretty sure the complaints are less about the template and more about the article's inclusion in WP:LAME, plus it looks like the template won't be deleted. Personally, I don't think the actual lame edit war for the "Nobel Prize in Economics" article is the one listed in WP:LAME -- there are others that could be put there, but I won't add them since the edit war isn't over and it would probably only exasperate the situation. –panda 15:05, 15 November 2007 (UTC)[reply]
I would like to invite you to take a look at the discussion happening on the Talk:1 E-3 s. You and I are heading towards a slipery slope of WP:3R. Surelly you can explain in better details and provide some sources for your mis-information. (per WP:V the accronym you have provide does not meet the standards for inclusion)... I'm copying this message to the talk:1 E-3 s. --CyclePat (talk) 18:42, 19 November 2007 (UTC)[reply]
After looking at the history of 1 e-3 s I have noticed that you violated the 3R rule. This is because:
Hello Itub, I would first like to apologize for all the comotion at talk:millisecond. I've left a reply on the talk page for you. Best regards, in all humility, Thank you. --CyclePat (talk) 19:09, 21 November 2007 (UTC)[reply]
Hey there! Redyva has loved you by placing a heart icon in the top-right corner of your userpage. Don't worry, it's not vandalism, but simply a small way to spread the WikiLove. If you don't really like it, feel free to revert it and make it go away, and no hard feelings; after all, it's just a small token of appreciation. If you like it, just add your name here, but again, there's no need to feel upset if you don't. Love and best wishes, Master Redyva (talk) 19:47, 21 November 2007 (UTC)[reply]
In the future, please resort to the discussion page, before you try to edit something you think is wrong. Or it could result in a wikiwar which is against the rules.
There are two kinds of oxidation states: Formal oxiodation sates and averaged oxidation states.
Take the permanganate ion for instance, in it, Manganese has a formal oxidation state of +7. There can be be no indiscrepencies.
Now, take the N2F2 molecule, because the two nitrogen atoms are bonded together, they form a completely covalent radicle concisting of N22+. In this radicle the 2+ oxidation state is shared between the participants.
This makes the 1+ simply a hypothetical average oxidation state if you were to isolate one nitrogen from the other. It is a calculated illusion, nothing more, it does not exist in the N2F2 molecule.--222.153.150.124 (talk) 21:02, 10 December 2007 (UTC)[reply]
Nitrogen does form compounds in the +1 oxidation state (FN=NF) and in the -1 oxidation state (HN=NH). I've reverted your edit accordingly. Please look at a decent book such as Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN978-0-08-037941-8. before deleting more oxidation states. --Itub (talk) 12:29, 10 December 2007 (UTC)[reply]
Why do you think that Carbon has a -1 oxidation state. If your trying to use the carbide ion as an example, I should point out that it is the same case as in N2F2 in Nitrogen. The "-1" state does not occur anywhere out side of the C22- radicle.--Plasmic Physics (talk) 21:22, 10 December 2007 (UTC)[reply]
The -1 oxidation state occurs in acetylene, in carbon 1 of ethanol, ethylamine, chloroethane, and all other similar compounds with a C(CH2)X substructure where X is an atom more electronegative than carbon. It also occurs in the central carbon of isobutane. And many, many more compounds. Read oxidation state. And read the Chemistry of the Elements by Greenwood and Earnshaw. You'll see that their table of oxidation states lists -4, -3, -2, -1, 0, 1, 2, 3, and 4 for carbon, and -3, -2, -1, 0, 1, 2, 3, 4, and 5 for nitrogen. --Itub (talk) 07:14, 11 December 2007 (UTC)[reply]
BTW, C22- is not a radical (let alone a radicle!). It is an anion. --Itub (talk) 07:16, 11 December 2007 (UTC)[reply]
Terminology! My mistake, I meant anion. Two atoms or ions that both have positive or negative oxidation states are not and cannot be bonded or ionically or otherwise. Negative to positive, neutral to neutral, or negative to neutral to positive, not negative to negative, or positive to positive. Just because one book cites information doesn't mean its true. If Nitrogen in dinitrogen difluoride has a formal oxidation state of +1 then according to basic physical chemisty rules, the nitrogen cannot be bonded to the other, as I just explained.Plasmic Physics (talk) 07:57, 11 December 2007 (UTC)[reply]
You are confusing oxidation state with charge. Atoms with the same oxidation state can and do bond to each other. What part of the definition of oxidation state you don't understand? It is a hypothetical charge that an atom would have if the electrons were partitioned according to an arbitrary set of rules (the electrons in a bond are "counted" as belonging to the most electronegative atom). In any case, even atoms with partial charges of the same sign can form covalent bonds to each other too. Chemical bonds are not simple electrostatic attraction. --Itub (talk) 08:41, 11 December 2007 (UTC)[reply]
I know that oxidation state is related but not the same as charge. If your theory is correct, then why is the (I) only stable in the dimer form (N22+), why does this collapse into a different state outside of the dimer? You don't get the NF molecule.
If a 1/2 is a formal oxidation state of oxygen then why does no molecule exist with that atom that doesn't contain the dioxygenyl cation?Plasmic Physics (talk) 10:09, 12 December 2007 (UTC)[reply]
Both arguments are non-sequiturs. Oxidation states can and are defined for atoms in molecules and don't need to be stable "by themselves", whatever that means. --Itub (talk) 10:51, 12 December 2007 (UTC)[reply]
Its time to reach common ground, here is my suggestion When we add new oxidation states, they must:
Provide an refference to an example.
Occur in inorganic compounds.
Occur unbonded to another atom of the same element with the same oxidation state in the example.
If you genuinely feel that you have a new oxidation state but, it does not comply with these rules, then add it to the main article not the info box. And state the conditions under which that oxidation state occurs.Plasmic Physics (talk) 22:40, 12 December 2007 (UTC)[reply]
I'm sorry, but I can't agree with those suggestions. I see no reason for excluding organic compounds and compounds with homonuclear bonds. There's nothing special about these compounds and nothing exotic about those oxidation states. --Itub (talk) 09:15, 13 December 2007 (UTC)[reply]
OK, so this is a matter of ego, since either way your oxidation states could be introduced into the article? Do we need to bring in someone more supperior, perhaps an auditor? Do you not want to put an end to this incessant argumentation?Plasmic Physics (talk) 10:25, 13 December 2007 (UTC)[reply]
Thanks for removing "whistogen" and "dracogen" which I had wondered about as well. I also question the same editor's group numbers for the lanthanides: IC to VIIIC with seven (!) elements numbered VIIIC. Certainly Mendeleev and Bohr would be surprised, but I would like your opinion. Have you ever seen these numbers before, or are they equally nonsense which should be deleted? Dirac66 (talk) 16:04, 12 December 2007 (UTC)[reply]
I have never heard of these group numbers, but I decided to give them the benefit of the doubt until I have time to look at it more carefully. --Itub (talk) 17:08, 12 December 2007 (UTC)[reply]
I decided to remove them too. They seem extremely dubious and I haven't found any plausible google hits for them. --Itub (talk) 17:14, 12 December 2007 (UTC)[reply]
Good decision, and your edit summary is well expressed. Dirac66 (talk) 20:11, 12 December 2007 (UTC)[reply]
Its acctually quite sensible to have seven VIIIC groups, it is no more senseless than having three VIIIB groups. Section A has one, if group 0 is concidered as group VIIIA, section B has three, section C has seven, and the g-block would have eleven. It would be like saying that Earth has one moon, why should Jupiter have more moons than Ganymede.Plasmic Physics (talk) 23:02, 12 December 2007 (UTC)[reply]
Answer to Plasmic Physics:
Your group numbering may seem sensible to you, but it is not the numbering used by any chemistry book or article that I have ever seen, or that Itub has ever seen according to his answer above. Wikipedia policy is to summarize information which is accepted elsewhere, not to provide a forum for everyone's personal unpublished ideas on better ways to do things such as numbering the groups of the periodic table. For more details on this policy, see WP:No original research. Dirac66 (talk) 01:57, 13 December 2007 (UTC)[reply]
Right, I will comply, but as a final note, it was not my own research, I just lack the ability to accurately refference it.Plasmic Physics (talk) 08:03, 13 December 2007 (UTC)[reply]
Hi there, I've made some chages to the article and replied to your comments on the FAC page. Could you have a look and see if I have dealt with your concerns? Tim Vickers (talk) 10:45, 21 December 2007 (UTC)[reply]

 J O R D A N [talk ] 16:08, 15 March 2007 (UTC)
J O R D A N [talk ] 16:08, 15 March 2007 (UTC)



![{\displaystyle K{\stackrel {\mathrm {def} }{=}}{\frac {\left[C\right]^{c}\left[D\right]^{d}}{\left[A\right]^{a}\left[B\right]^{b}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e86ea654d2126431379665baa730e369eb05edf6)








{kind=link}